Current Research

- In February 2010, the results of the MermaiHD study were announced as a press release: pridopidine, a dopamine stabiliser (also known as Huntexil, previously known as ACR16), was shown to improve voluntary motor symptoms in a phase lll trial of 437 patients with Huntington’s disease. Publication in a peer reviewed form is awaited.
- Previous clinical trials have evaluated creatine and coenzyme Q10 among other compounds, but have not demonstrated efficacy. Many patients continue to buy them over the counter and comparison trials of the two substances, funded by National Institutes of Health, are ongoing.
- A phase lll trial of latrepirdine (Dimebon) is currently taking place in multiple sites, including several in the UK.
- Enhancing clearance of mutant huntington by cellular clearance mechanisms: several compounds being tested in mouse models of Huntington’s disease aim to promote clearance of the mutant protein, huntingtin, which is generated by the expanded HTT gene
- Histone deacetylase inhibitors: these target the transcriptional dysregulation that occurs early in the pathogenesis of Huntington’s disease.
- Inhibitors of proteolytic cleavage of full length mutant huntingtin: these would prevent production of the potentially toxic N-terminal fragment
- Gene silencing: to switch off expression of the mutant gene itself
Much progress has been made in developing and evaluating sensitive biomarkers that will help measure the effects of disease modifying treatments in future clinical trials, particularly in the premanifest and early stages of the disease. Track-HD and Predict-HD are major international collaborative studies that have increased our understanding.
Latest Huntington’s disease research news, provided by HDbuzz.
Links
A new way of thinking about trials to prevent Huntington's disease Can we test drugs to delay or prevent the onset of Huntington's disease? New research suggests it's possible
Update confirms Huntington's disease 'gene silencing' trial on track Ionis says its trial of HTTRx, intended to lower huntingtin protein, is fully recruited and plans to extend it
New roles for huntingtin: removing a healthy protein to understand its function Completely removing normal huntingtin in adults may disrupt healthy brain function, a recent study suggests.
Precision huntingtin-lowering drug trials target the mutant protein WAVE Life Sciences launches PRECISION clinical trial to suppress the mutant Huntington's disease protein
A step forward for gene editing: CRISPR-Cas9 and HD Evolving CRISPR-Cas9 techniques can now be used to edit the HD gene in a living mouse brain.
New study reveals a potential HD biomarker A potential HD biomarker has been uncovered in a recent clinical study
Has a "wonder drug" for dementia been discovered? (Spoiler alert: no.) Media reports of a wonder drug for neurodegenerative diseases like Huntington's disease are overhyped
Huntington's Disease Therapeutics Conference 2017 - Day 3 HDBuzz summarises final day of the 2017 Huntington's Disease Therapeutics Conference in Malta
Useful websites for current research information
www.en.hdbuzz.netwww.ninds.nih.gov
www.youramazingbrain.org/huntingtonnew.htm
www.sciencedaily.com/news/mind_brain/huntingtons_disease/
www.native.com/subjects
www.newsciences.ucsd.edu/centers/huntingtons-disease/pages/default-cspx
www.newshd.net
Breakthrough drug in the treatment of Huntington’s Disease!
Huntington’s disease (HD) is a devastating neurodegenerative disease. Patient’s brain cells are in decline and this leads symptoms such as: memory loss, mood changes, jerky movements and results in death. The cause of this terrible disease comes from an error in the Huntington’s gene, a CAG DNA repeat that is much longer in HD patients than in people without the disease. This fault is carried in the messenger RNA, which in turn leads to the creation of a faulty Huntington’s protein. Source: http://www.bbc.com/news/health-42308341
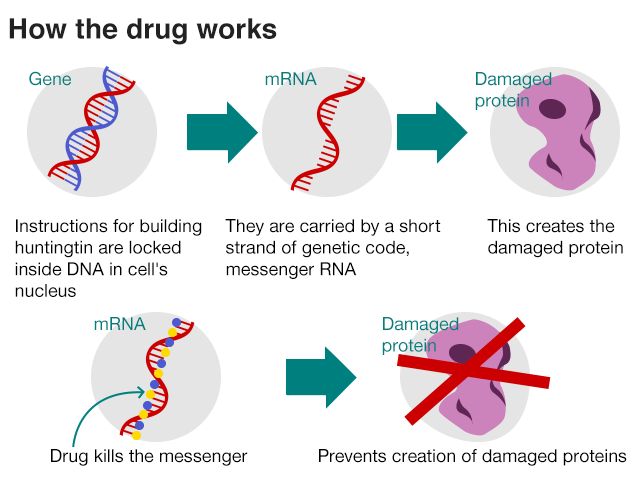
The trial included 46 patients who were injected with the drug, in the spinal fluid. Researchers then looked at whether the drug was safe and whether it lowered the amount of faulty protein in the brain. Their findings follow on from the mouse study where they found that by lowering the amount of mutant Huntington’s protein motor functions recovered.
The drug now needs to go through phase 3 clinical studies, which will further establish the safety of the drug and also look at the effects on the drug, on the symptoms of Huntington’s disease such as motor functions.
This has the potential to open the doors for treatments into other neurodegenerative diseases such as Parkinson’s and Alzheimer’s.

Definitions
DNA is an acid in the chromosomes in the centre of the cells of living things. DNA determines the particular structure and functions of every cell and is responsible for characteristics being passed on from parents to their children. DNA is an abbreviation for 'deoxyribonucleic acid'.
RNA is an acid in the chromosomes of the cells of living things which plays an important part in passing information about protein structure between different cells. RNA is an abbreviation for 'ribonucleic acid'.
CAG repeat diseases: A group of neurodegenerative diseases characterised by the repetition of the nucleotides cytosine-adenine-guanine in-specific genes. Diseases in this group include Huntington’s disease.
For more see:
HD Research Update
August2018
Anotherstep forward in the continuing fight to defeat this catastrophic disease
IONIS-HTTRx (RG6042) Granted PRIME Designation by the European Medicines Agency for the Treatmentof People with Huntington's disease. IONIS-HTTRx is the first and only drug to demonstrate reduction of mutant huntingtinprotein, the underlying cause of Huntington's disease, in patients. Roche plans to initiate a pivotal study ofIONIS-HTT Rx (RG6042).
PRIME (PRIority Medicines) designationallows the drugs testing timeline to be sped up and the EMA will prioritiseassessment of the drug. Thisdesignation is usually given to drugs for diseases such as HD, where there areno available treatments.
"PRIMEdesignation for IONIS-HTTRx accelerates the review timelines and enhancesinteractions with the EMA, which can bring this potentially disease-modifyingdrug for people with Huntington's disease to regulatory approval faster. Thisdesignation will be useful as we work closely with Roche to quickly advanceIONIS-HTTRx into a pivotal study," said Dr. C. Frank Bennett, senior vicepresident of research and franchise leader for the neurological programs atIonis Pharmaceuticals. "This is our second antisense drug to demonstrate astrong safety profile and significant target engagement in the human centralnervous system. This profile gives us further confidence in the potential ofthe many other drugs we have advancing in R&D for the treatment ofneurological diseases."
TheFDA has also granted IONIS-HTTRx orphan drug designation. This is the USequivalent of the PRIME designation and again is for drugs that are treatingdiseases where there is no treatment.
ABOUTIONIS-HTTRx (RG6042)
“IONIS-HTTRx(RG6042) is an antisense drug designed to reduce the production of all forms ofthe huntingtin protein (HTT).” In particular it targets the mutant protein form(mHTT), accumulation of which leads to HD.
“In a Phase 1/2 study, IONIS-HTTRx (RG6042) demonstrateda significant reduction in mHTT, which breaks down the nerve cells in thebrain. The study demonstrated a mean 40% (up to 60%) reduction of the specificHD protein in the cerebrospinal fluid (CSF) of adult patients treated withIONIS-HTTRx (RG6042) for three months at the two highest doses. Furthermore,levels of mHTT measured in the CSF were still declining in the majority oftreated patients (~70%) as of the last measurement in the study. IONIS-HTTRx (RG6042) was well tolerated in thisstudy.”
Phase 3 will involve long termstudies which will look at patient safety and side effects in a larger patientpopulation. For more information:
The fight, with your help, goes on to win the battle against HD
Useful websites for more information:
Latest News on HD Project – WAVE

Dear global HD community,Today we announce encouraging top line results from Precision – HD2, our ongoing Phase 1b/2a placebo-controlled trial evaluating the investigational therapy WVE- 120102 targeting SNP2. In an analysis of the study results, we compared all patients treated with WVE-120102 to patients who received placebo and saw a statistically significant reduction of 12.4% (p<0.05) in mutant huntingtin protein in the cerebrospinal fluid (CSF).
In addition to demonstrating a reduction in mutant huntingtin protein, WVE- 120102 was generally safe and well tolerated across all patients, so we are exploring optimal dosing and expect to add an additional treatment group to the ongoing Phase 1b/2a study at a higher dose (32mg) in January to evaluate the effects on the mutant huntingtin protein. We are also adding an additional treatment group to our Phase 1b/2a called PRECISION-HD1, evaluating the investigational therapy WVE-120101 targeting SNP 1. We now expect to share results from both studies in the second half of 2020.
We are deeply committed to the HD community. In addition to the PRECISION –HD studies, we are developing other potential treatments for the disease, advancing the understanding of wild-type huntingtin preservation, and supporting the needs of people living with HD.
All of us at WAVE are enormously grateful to the PRECISION-HD clinical trial participants and their families. We recognise the personal sacrifices made by each and every family involved in these trials. Their participation along with the support of the entire HD community are critical to advancing the scientific and medical understanding required to defeat this devastating disease.
What is the PRECISION-HD program?
Our PRECISION-HD program is the first clinical program to use an allele-selective approach to target the underlying cause of Huntington’s disease. PRECISION-HD1 and PRECISION-HD2 are Phase 1b/2a multicenter, randomized, double-blind, placebo-controlled clinical trials evaluating the safety and tolerability of WVE-120101 and WVE-120102, respectively.
Will you initiate a Phase 3 clinical trial of WVE-120102?
Our current focus is on delivering the 32 mg dataset in the second half of 2020.
We would anticipate working on continued development of WVE-120102 including Phase 3 studies if the data from the ongoing study continue to support it.
How can I enroll in the new treatment groups for the PRECISION-HD trials?
We expect to enroll patients into the 32 mg treatment group primarily at sites that participated in the multi-dose portion of the Phase 1b/2a PRECISION-HD2 study, which only enrolled patients outside of the U.S. If you have questions about participating in a clinical trial we encourage you to speak directly with your physician.
What happens now to the patients who are in the trial, what dose are they being given?
In October 2019, we initiated an open-label extension (OLE) study, which is open to patients outside the United States. Initially patients will continue on their existing doses except for the 2 mg cohort where they will receive 4 mg.
Latest News on Huntington’s Research
October 2020 - Branaplam
While developing a drug called branaplam for patients with SMA, the pharmaceutical company Novartis discovered that it could hold promise for people with HD. The FDA has granted a special status called Orphan Drug Designation to branaplam.
Branaplam was originally developed for the treatment of the neurological disorder spinal muscular atrophy (SMA) and is still under clinical investigation for this purpose. Branaplam works by interfering with how genetic messages are processed in cells and can increase the levels of a protein called SMN2. Boosting levels of SMN2 helps SMA patients who have lower levels of this protein which is the underlying cause of the disease in many cases.
Interestingly, it seems that branaplam also reduces the levels of the huntingtin gene message and the huntingtin protein. This is what huntingtin-lowering therapies all aim to do and is how many researchers and companies hope to treat Huntington’s disease.
The huntingtin-lowering therapies currently in clinical trials need to be given through spinal injections or brain surgery. Unlike these, branaplam is a small molecule drug, which means it can be taken as a tablet. If Novartis can show that branaplam is an effective treatment for HD, this would mean that it could be given as a pill, a more convenient option for HD patients!
February 2021 – GPR52
Scientists have found new leads for how to reduce the levels of the Huntington’s disease (HD) protein by targeting a protein called GPR52. A team of researchers working in Shanghai, China developed small drug-like molecules which lower huntingtin protein levels in HD tissue culture models and in HD mouse models. Treatment with their molecules was also shown to improve the HD mouse symptoms.
New avenues for huntingtin lowering
Huntingtin-lowering therapies are heralded by many researchers and clinicians as a very promising avenue for treatment of HD. People with HD have a longer or expanded CAG number in their HD gene, huntingtin. This means that throughout the body and brain they make a different, longer form of the huntingtin protein. This expanded protein is thought to be responsible for many of the symptoms we observe in people with HD.
The idea of huntingtin-lowering therapies is that if you reduce expanded huntingtin, you might also get rid of its toxic effects. There is evidence in lab HD models that huntingtin-lowering can slow or improve HD symptoms, but we await the results of ongoing clinical trials before we can be sure that the same effect will be observed in patients.
What has GPR52 got to do with huntingtin lowering?
GPR52 is part of a family of proteins called G-protein coupled receptors that sit on top of cells and receive messages, similar to a satellite dish. These types of receptors are commonly targeted by FDA-approved drugs. This gives scientists optimism that using new medicines to target members of this receptor family, like GPR52, could be safe and effective.
GPR52 was first identified by scientists as a protein of interest for HD in a genetic screen. A genetic screen can be thought of as a scavenger hunt, where scientists look at different genes one-by-one to see how they might, in this case, change signs of HD. Scientists then used genetic tricks in the lab to delete the GPR52 gene or to lower the levels of GPR52 protein, and this helped to improve signs of HD in cells and flies. A team of researchers in Shanghai, China, recently took this one step further and tried reducing the levels of GPR52 in mice. In this recently published research, they show that reducing GPR52 reduces the levels of huntingtin, indicating that GPR52 would be a good candidate to target with small molecules for huntingtin-lowering therapies.
Targeting GPR52 with small molecules lowers huntingtin and improves symptoms in HD lab models
The scientists describe the development of small molecules which stick on very tightly to GPR52 but not other proteins in our cells. These type of properties of a small molecule suggest that down the road this potential therapy could be very specific and avoid other unwanted effects. The best or lead molecule identified in this study, called Comp-43, was then used in a variety of different experiments to test how well it worked for huntingtin-lowering and reducing signs of HD-related damage and improving behaviour.
They first showed that Comp-43 could reduce huntingtin levels and preserve the health of mouse nerve cells in a dish. Following this exciting result, the scientists went on to test Comp-43 in HD mice. Critically, Comp-43 could cross the blood brain barrier in mouse models and lower huntingtin levels in some brain regions. This suggests that if Comp-43 was developed into a medicine in the future, people with HD might take it as a pill. Mice treated with Comp-43 had improved symptoms when tested on laboratory tasks that measure their movement and coordination, like a log-rolling exercise called the Rotarod or their ability to cross a balance beam. They also showed that mice treated with Comp-43 had more and healthier brain cells.
So, what’s next for GPR52 in HD research?
All of the data available in the literature to date indicates that GPR52 is a very promising drug target for huntingtin-lowering therapies but there are still many hurdles to cross before we are treating patients with GPR52 targeting molecules. Many therapies work well treating HD in cells in a lab dish or in different animal models of HD but don’t necessarily go the distance in safely treating people with HD.
It is not yet apparent what additional effects targeting GPR52 might have, beyond changing huntingtin levels and HD symptoms. GPR52 has important jobs to do in our nervous system and may play a role in dopamine signalling, chemical messages that affect mood, movement, and motivation. Sustained targeting on GPR52 by this type of huntingtin-lowering therapy might have side effects which we just haven’t yet observed in the shorter treatment experiments completed to date.
It should also be noted that targeting GPR52 lowers the levels of both normal and expanded huntingtin. We know that targeting both forms of the protein with other therapies such as Roche’s tominersen, which is also non-selective, is safe in the trials completed to date, but targeting expanded huntingtin while preserving normal huntingtin is still the goal for many researchers.
Nonetheless, small molecule targeting of GPR52 represents an exciting new way to lower huntingtin levels and we expect to see answers to many of these outstanding questions in future studies.
March 2021 - Tominersin
Very sad news was announced today as Roche and Ionis declared that the large ASO study they’re running in Huntington’s disease patients has been halted early. Importantly, no specific new safety concerns were raised so far, but nevertheless dosing of the study drug, tominersen, as well as the placebo, has been stopped prematurely. What does this mean, and where do we go from here?
Background - what’s this trial doing?
Roche and Ionis have developed tominersen which is a type of drug called an antisense oligonucleotide, more commonly referred to as an ASO. ASO therapies are able to reduce the levels of specific protein molecules by interfering with the genetic message which normally tells the cells of our bodies to make that protein. In the case of tominersen, this ASO drug interferes with the huntingtin protein genetic message. Tominersen treatment lowers the levels of both regular and Huntington’s disease versions of the huntingtin protein.
This is not the first tominersen clinical trial. Before getting to this phase III study, tominersen was first tested in a Phase I/II trial where it was assessed for safety and shown to lower the levels of huntingtin protein in HD patients. The aim of this current phase III trial, also called the GENERATION-HD1 trial, was to work out if tominersen was effective, not only at lowering huntingtin protein in a larger group of patients, but also if it helped improve signs of HD in patients already showing symptoms.
What happened?
On March 22nd, 2021, a press release from Roche revealed that the phase III study of tominersen had halted dosing on the advice of the Independent Data Monitoring Committee (iDMC). This committee is a group of independent experts who have been monitoring the data from the ongoing study since it began.
These data monitoring committees play a very important role in clinical trials - their job is to act as a neutral party by looking at the data emerging from the trial, without an interest in the trial outcome. So, by design, these committees are totally separate from the patients, physicians and drug companies involved in running the study. Their sole job is to monitor the trial periodically to determine whether or not it should continue.
In general, these committees are asking two kinds of questions - first, are there any unexpected safety concerns emerging? If, for example - all the people receiving a drug had started to have some very weird symptom, this committee would see that and order the trial halted. Secondly, these committees can determine whether ongoing trials look extremely unlikely to have any benefit for the patient participating.
For example, earlier HD studies of several drugs have been halted by this kind of early analysis because the data suggest that the patients are extremely unlikely to benefit from the drug. If folks’s HD symptoms are clearly not getting better, then the risk/benefit calculation for giving experimental drugs to people has shifted, and it may no longer be worth it to continue the trial.
What do we know?
What we know is very limited, which is important to remember for the next few weeks and months. All the important facts we know from the press release come from these few sentences:
“The decision was based on the results of a pre-planned review of the data from the Phase III study conducted by an unblinded Independent Data Monitoring Committee (iDMC). The iDMC made its recommendation based on the investigational therapy’s potential benefit/risk profile for study participants. No new or emerging safety signals were identified for tominersen in the review of the data from this study.”
That tells us a couple things. First, that there’s no new “safety signals” - meaning no new bad medical outcomes for the people in the trial. If there had been, say, sudden heart attacks in patients getting the drug (there weren’t!!), this press release would have to tell us that. So, thankfully for the families participating, there’s not yet any sign of scary new symptoms that emerged.
Second, the press release says that they decided to halt dosing in the trial because of the “investigational therapy’s potential benefit/risk profile for study participants.” So how could the benefit/risk profile have changed if there’s not scary new symptoms that emerged? Bottom line - we don’t know yet. However, hypothetically speaking, it could be that the drugs worsen HD symptoms. Or, hypothetically, it could be that the drug just doesn’t make HD symptoms better, in a way that is obvious to someone who has access to all of the data, and so it’s not worth the risk of exposing people to a new drug because the benefits aren’t clear.
Importantly - not even the researchers at Roche and Ionis know the answer to those hypothetical questions right now. When things like this happen, the independent committees have to make their decisions and let everyone know at once. So, until you see another HDBuzz story about this, tune out any noise you hear about these results - there’s a huge amount we just don’t know.
What do we not know, and when will we know it?
Why have we gotten this cryptic press release for something so important? Unfortunately, the way this works is that when trials are halted like this, there’s an initial press release that’s released as soon as the companies found out about the news. This is both so that the patient community can be informed, and to prevent shenanigans with people learning the news and selling stocks, or other inappropriate things.
As an example, after today’s news Ionis’s stock fell by nearly 19%, representing more than a billion dollars worth of value lost. In the wrong hands, early access to this information could be mis-used.
For the patient community, this means that when we hear bad news like this, there’s always a baffling gap between this initial warning that something hasn’t gone right, and learning the details of exactly what’s happened. This is incredibly frustrating, but just how it goes when we hear things like this.
Is HTT lowering a bad idea?
We’re likely to see a lot of discussion - and rightly so - about whether lowering huntingtin protein is a bad idea. Based on all the science we had at the time, we believed it was right to run this trial. And we have this incredible new set-point, which is that we know we can lower huntingtin protein in HD patients. So when we design the next trials - and there will be more trials - we’re not going back to square one, but rather a point at which we know that lowering huntingtin in patients is possible.
What’s next?
Until we see the data, there’s no way to predict what will happen next. But here are some ideas that are sure to be discussed: first, should we be trying to treat HD patients earlier, even before they have advanced symptoms? Second, should we be trying to only lower mutant huntingtin protein (as Wave Life Sciences is currently trying to do in their ongoing study)? Third, should we be trying to lower huntingtin more or less than we did in this trial? You can bet that folks at Ionis and Roche are talking about this right now, as are other HD scientists around the world.
Meanwhile, even though dosing has been permanently stopped in the GENERATION-HD1 trial, Roche intends to continue monitoring participants to measure the longer term safety and effects of tominersen.
Gratitude for patients, physicians and companies
This trial was an enormous undertaking. The HD patients who volunteered in these early trials are forever HD heroes who took on significant risk for themselves on behalf of the entire HD community. Researchers in basic science labs, at both Ionis and at Roche, worked tirelessly to design the best possible drug for testing. And everyone who worked in clinics around the world to test the drug labored relentlessly to see if the drug worked. Everyone involved - families, scientists and physicians - wanted another outcome, but we just didn’t get it this time.
Update – HD Therapeutics conference
Roche presented their data from the failed clinical trial, showing that at the doses and time points looked at, the ASO had no positive effect on patients- in comparison to a placebo. Basically, their symptoms did not improve because of the dosing, in fact at one dosing level, symptoms seemed to get worse. Whilst we do not fully understand the implications of this for future trials, Roche has committed to sharing the data to help improve trial design and HD drug research. This has been disappointing for the HD community, however, there is a lot of exciting research in the pipeline that will benefit from all the data gathered.
Email address: info@addor.co.uk
Please click the image below to donate by bacs
Logo Design: Iona Page
Thomas Francis O’Reilly
Timothy Nigel O’Reilly
Andrew Kevin O’Reilly
Fourth Trustee to be advised
HD Support & Technical Advisor
Daniel Timothy O’Reilly
Privacy & Data Protection Statement